L'année 2022 a marqué les 50 ans du dépistage néonatal en France. Jusqu’ici cet examen préventif permettait de dépister 6 maladies rares ainsi que la surdité permanente néonatale.

L’année 2022 a marqué les 50 ans du dépistage néonatal en France.
Jusqu’ici cet examen préventif permettait de dépister 6 maladies rares ainsi que la surdité permanente néonatale.
Le dépistage néonatal, c’est quoi ?
Le programme national de dépistage néonatal (DNN) est une démarche de santé publique. Il concerne tous les nouveau-nés qui naissent en France mais existe aussi dans de nombreux pays étrangers. Le but est de prévenir la survenue de manifestations et de complications graves dues aux maladies dépistées, a minima d’en limiter la gravité. Parce qu’il existe des moyens thérapeutiques préventifs, le dépistage précoce apporte un véritable bénéfice aux nouveau-nés.
Son objectif est de dépister certaines maladies rares, et de prendre en charge le bébé avant même l’apparition des premiers signes, pour lui permettre de se développer sans séquelles et de grandir le mieux possible.
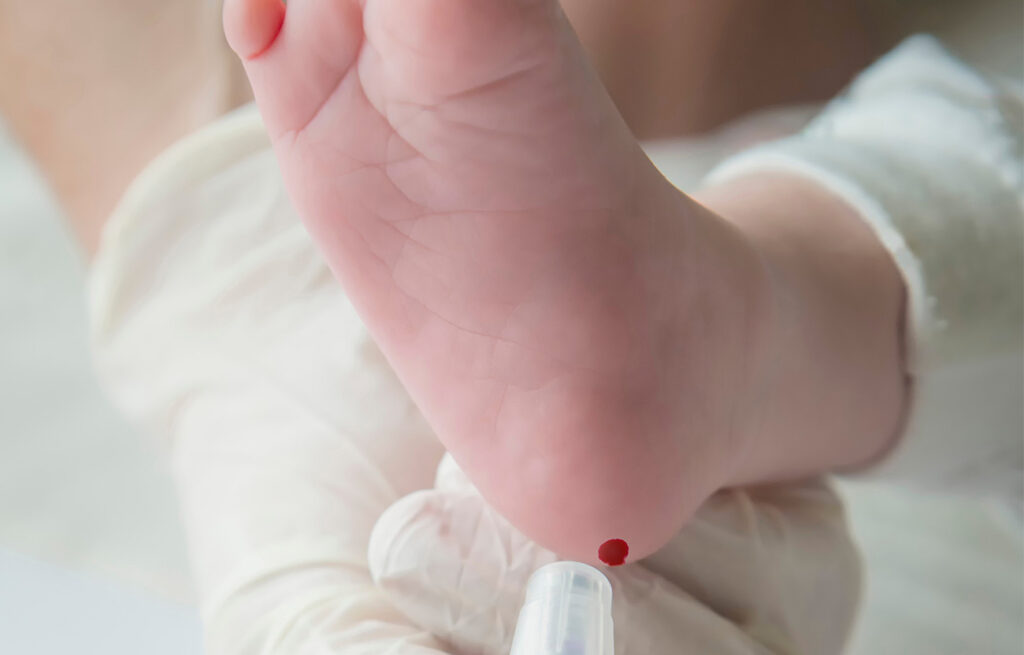
Le dépistage néonatal est réalisé en prélevant des gouttes de sang sur un buvard, après une petite piqûre au talon du nouveau-né. Il est systématiquement proposé et réalisé après accord des parents. Réalisé gratuitement, le prélèvement est fait le plus souvent en maternité – parfois au domicile – dans les 3 jours suivant la naissance.
Les résultats ne sont communiqués aux parents qu’en cas de problème.
Les maladies actuellement dépistées à la naissance sont :
- La phénylcétonurie : maladie génétique due au déficit d’une enzyme qui transforme la phénylalanine présente dans l’alimentation. En l’absence de traitement, elle peut entraîner un retard mental sévère et des complications neuropsychiatriques ;
- L’hypothyroïdie congénitale : maladie qui se traduit par une sécrétion insuffisante des hormones thyroïdiennes par la glande thyroïde. En l’absence de traitement, son dysfonctionnement retentit sur les grandes fonctions de l’organisme et peut avoir notamment pour conséquence un retard mental sévère ;
- L’hyperplasie congénitale des surrénales : défaut génétique du fonctionnement des glandes surrénales. En l’absence de traitement, elle peut être à l’origine de déshydratations aiguës sévères, parfois mortelles, et de troubles du développement des organes sexuels ;
- La mucoviscidose : maladie génétique qui entraîne des infections respiratoires sévères et répétées ainsi que des complications digestives ;
- Le déficit en MCAD (Medium-Chain-Acyl-CoA Déshydrogénase) : maladie qui entraîne une difficulté de l’organisme à utiliser les graisses comme source d’énergie. En l’absence de traitement, elle peut provoquer des comas pouvant aller jusqu’au décès de l’enfant ;
- La drépanocytose, si l’enfant fait partie des populations exposées à cette maladie : La Haute Autorité de santé (HAS) vient de publier un avis recommandant la généralisation du dépistage de la drépanocytose sur l’ensemble du territoire national. Il était, jusqu’alors, le seul dépistage à être ciblé, c’est-à-dire qu’il visait à identifier en priorité les populations les plus à risque de développer une drépanocytose. Une prise en charge le plus tôt possible pour cette pathologie permet d’éviter de graves répercussions en santé et améliore la qualité de vie du patient. Cette pathologie est la plus fréquente parmi les maladies génétiques initialement dépistées en France, et une augmentation du nombre de cas a été constatée au cours de la dernière décennie (plus de 50% d’augmentation entre 2010 et 2020). Cette maladie génétique est liée à la présence d’une hémoglobine anormale dans le sang qui peut se traduire par une anémie persistante, des complications vasculaires, des crises douloureuses et des infections répétées.
- La surdité permanente néonatale.
Depuis le 1er janvier 2023, le programme national de dépistage néonatal est étendu à sept autres maladies et erreurs innées du métabolisme :
- L’homocystinurie : maladie génétique liée au déficit d’une enzyme, la cystathionine bêta-synthase, qui entraîne l’accumulation d’homocystéine toxique pour l’organisme. En l’absence de traitement, elle peut entraîner une atteinte des yeux, du squelette, du système vasculaire, du système nerveux et parfois un retard du développement ;
- La leucinose ou maladie des urines à “odeur de sirop d’érable” : maladie génétique liée au déficit d’une enzyme, la « déshydrogénase des alpha-céto-acides à chaîne ramifiée ». En l’absence de traitement, elle se caractérise par l’apparition rapide de difficultés pour s’alimenter, un temps de sommeil trop prolongé, des vomissements puis des troubles neurologiques, des mouvements anormaux et une insuffisance respiratoire
- La tyrosinémie de type 1 : maladie génétique liée au déficit de l’enzyme hépatique, la fumaryl acétoacétate-hydrolase qui permet la transformation normale des protéines contenues dans les aliments. Sans un régime et un traitement appropriés, des déchets toxiques s’accumulent dans l’organisme et endommagent gravement le foie, les reins et le système nerveux ;
- L’acidurie isovalérique : maladie génétique, appelée également acidémie isovalérique, liée au déficit d’une enzyme, « l’isovaléryl-CoA déshydrogénase », responsable en l’absence d’un régime adapté de troubles aigus à la naissance (vomissements, convulsions) ou plus tardifs (retard de croissance et/ou de développement) ;
- L’acidurie glutarique de type 1 : maladie génétique liée à l’absence ou l’insuffisance de fonctionnement d’une enzyme, la glutaryl CoA-déshydrogénase. En l’absence d’un régime spécial, elle est à l’origine de l’accumulation de produits toxiques responsables de troubles neurologiques aigus chez les nourrissons ;
- Le déficit en 3-hydroxyacyl-coenzyme A déshydrogénase des acides gras à chaîne longue : maladie génétique liée à l’absence ou l’insuffisance de fonctionnement de cette enzyme. En l’absence de traitement et d’un régime adapté, elle se caractérise par la survenue dans la petite enfance d’une hypoglycémie, d’une atteinte au foie, de troubles cardiaques et neurologiques ;
- Le déficit primaire en carnitine : maladie génétique liée au déficit de transporteur de la carnitine. En l’absence d’administration de carnitine, ce déficit entraîne une atteinte cardiaque au début de l’enfance, souvent associée à une hypotonie, un retard de croissance, des crises hypoglycémiques récurrentes et/ou un coma.
Découvrez l'application 9moisetplus,
une équipe de sages-femmes disponible
pour vous répondre où que vous soyez dans le monde
Obtenez les réponses à vos questions rapidement.
Être écouté, conseillé et rassuré devient un jeu d’enfant.

